By
Niranjan Maharajh
November 5, 2025
•
20
min read
Over 580,000 bottles of prazosin hydrochloride capsules have been pulled from shelves across the United States following FDA enforcement actions against Teva Pharmaceuticals USA and Amerisource Health Services. The medications, used to treat hypertension and post-traumatic stress disorder, were found to contain elevated levels of N-nitrosamine impurities—potentially carcinogenic compounds that could increase cancer risk with prolonged exposure. This recall, announced in late October 2025, represents the latest chapter in an ongoing saga of nitrosamine contamination that pharmaceutical manufacturers and regulators have struggled to contain for nearly seven years. Yet for those monitoring industry trends and scientific literature, this crisis was not merely inevitable—it was predictable.
The recall encompasses three dosage strengths of prazosin hydrochloride capsules: 1 mg, 2 mg, and 5 mg formulations, with specific lot numbers and expiration dates spanning from 2025 through 2026. The affected products include approximately 181,659 bottles of 1 mg capsules, 291,512 bottles of 2 mg capsules, and 107,673 bottles of 5 mg capsules. The FDA classified this as a Class II recall, indicating that while the product "could lead to temporary or medically reversible adverse health effects," the immediate risk of serious harm is lower than a Class I recall. However, this classification may underestimate the risk to chronic users, particularly those taking prazosin for years to manage PTSD-related nightmares—a population potentially exposed to cumulative nitrosamine doses over extended periods.
The affected medication, prazosin, serves approximately two million Americans who take it for hypertension, with additional off-label use for PTSD nightmares and sleep disturbances. The sudden recall of multiple manufacturing lots creates disruption not only in patient continuity of care but also in healthcare provider confidence in pharmaceutical supply chain integrity.
The specific impurity flagged in the prazosin recall is identified as "N-Nitro Prazin impurity C," a N-nitrosamine compound that forms when secondary or tertiary amine functional groups within an active pharmaceutical ingredient (API) react with nitrosating agents under certain manufacturing or storage conditions. Prazosin, structurally speaking, is particularly vulnerable to this reaction. The drug functions as an alpha-1 adrenergic receptor agonist—a mechanism of action that requires a secondary amine functional group essential to its pharmacological activity. This structural feature positions prazosin squarely within the "cohort of concern" identified by pharmaceutical regulators for nitrosamine formation risk.
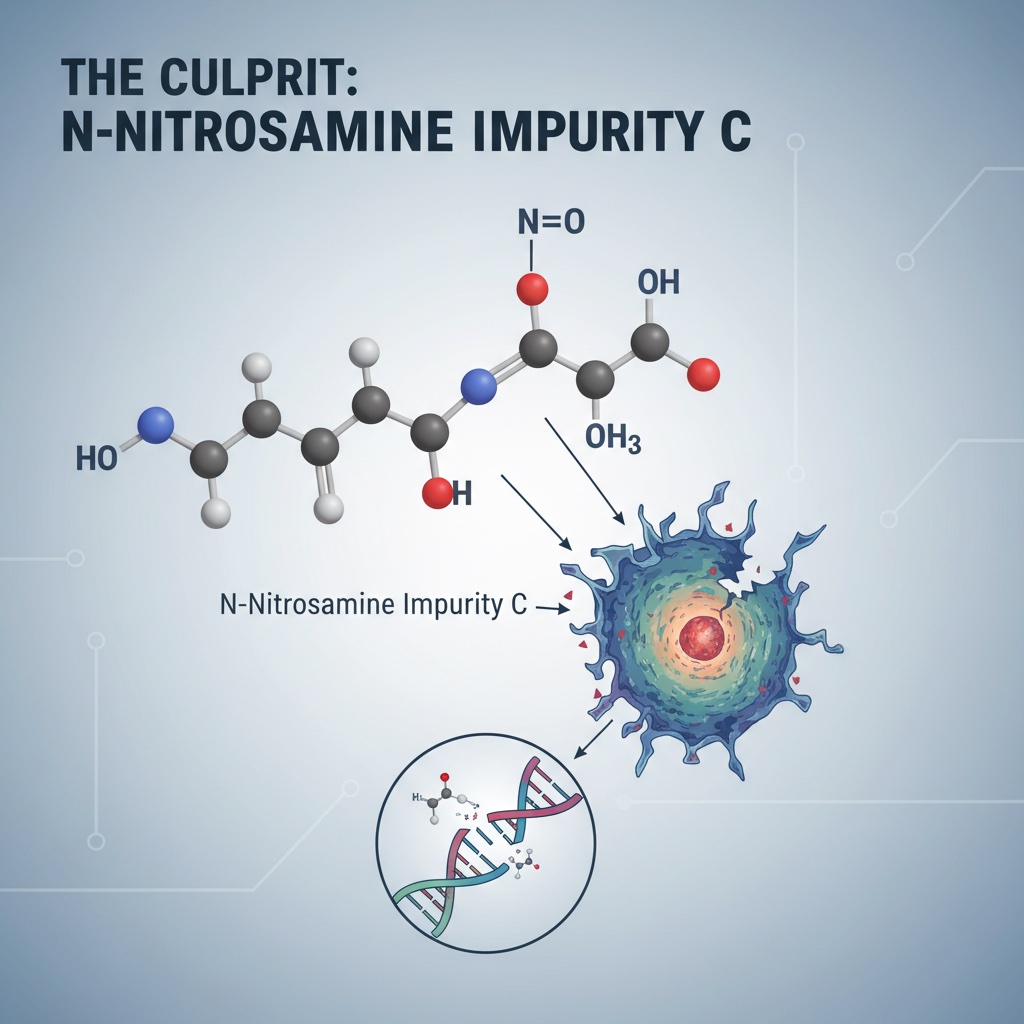
The presence of N-nitrosamines in medications has been classified by the International Agency for Research on Cancer as Group 2A compounds—"probably carcinogenic to humans." The FDA and European Medicines Agency have established acceptable daily intake (ADI) limits of 26.5 nanograms per day for most nitrosamines, with even lower thresholds for specific compounds like N-nitrosodimethylamine (NDMA) at 96 nanograms per day. When prazosin lots exceeded these thresholds, the regulatory machinery activated. Yet the question that should trouble manufacturers and regulators alike is simple: How did this slip through in the first place?
N-nitrosamine formation in pharmaceutical manufacturing is not mysterious or unpredictable—it requires three specific factors: (1) the presence of a nitrosatable amine, (2) the presence of a nitrosating agent, and (3) conditions conducive to N-nitrosamine formation. Prazosin satisfies the first requirement inherently. The second and third factors are controlled through manufacturing processes and material selection.
The documented root causes for nitrosamine formation in pharmaceutical production fall into several well-characterized categories:
Contaminated Raw Materials and Solvents: Trace nitrites are present in most common pharmaceutical excipients at various concentrations. Manufacturing suppliers may introduce nitrites through contaminated starting materials, intermediates, or recycled solvents. Dimethylformamide (DMF), commonly used as a solvent in pharmaceutical synthesis, can degrade into nitrosating species under certain conditions. Previous investigations of ARB recalls revealed that manufacturing process changes in 2014 involving sodium nitrite as a reagent led to NDMA formation—a cautionary tale suggesting that procedural modifications deserve heightened scrutiny.
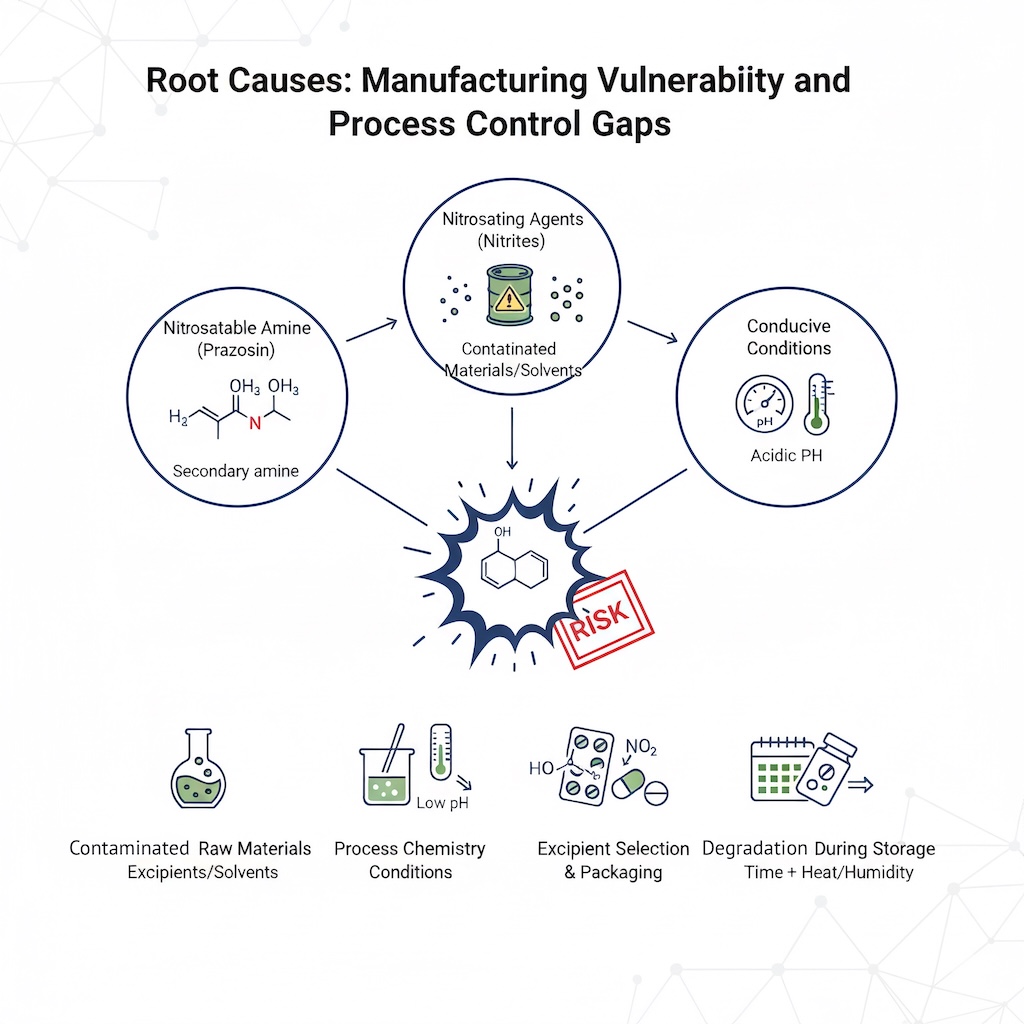
Process Chemistry Conditions: Acidic conditions dramatically accelerate nitrosamine formation. When wet granulation—a common pharmaceutical manufacturing step involving water—brings secondary amines into contact with trace nitrites in a mildly acidic environment, the chemistry becomes conducive to nitrosamine generation. pH control during manufacturing is therefore not a minor quality parameter; it is a critical risk mitigation factor.
Excipient Selection and Packaging: Excipients themselves can be nitrite sources. Crospovidone, a commonly used tablet binder, has been identified in multiple studies as containing elevated nitrite levels compared to alternatives like microcrystalline cellulose or lactose monohydrate. Additionally, primary packaging materials such as nitrocellulose-based blister packs can leach nitrite into the finished product during storage, particularly under elevated temperature and humidity conditions.
Degradation During Storage: Perhaps most insidiously, N-nitrosamines can form not during manufacturing but during the product's shelf life. Ranitidine, the H2-receptor antagonist recall of 2019, exemplified this mechanism: the drug degraded over time, generating secondary amine species that subsequently reacted with trace nitrites present in excipients. Crystal morphology and manufacturing process variations affected the rate and extent of this degradation—two variables that manufacturers may not have adequately characterized in their stability protocols.
For prazosin specifically, the FDA's classification of this as a "failure in the impurities and degradation specifications" suggests that Teva's manufacturing process likely failed to adequately control one or more of these factors—perhaps through insufficient monitoring of incoming material nitrite levels, inadequate pH control during formulation, or insufficient characterization of product degradation pathways during storage.
The irony of the prazosin recall is that it represents a fundamentally preventable failure. Since 2018, when nitrosamine contamination was first detected in valsartan (an angiotensin II receptor blocker), the pharmaceutical industry has had nearly seven years to implement preventive measures. The FDA issued comprehensive guidance in 2020 on controlling nitrosamine impurities, providing manufacturers with explicit risk assessment frameworks, analytical methods, and control strategies.
Effective prevention strategies include:
Comprehensive Risk Assessment Using ICH M7 Guidelines: The International Council for Harmonisation's M7 guidance provides a systematic approach for assessing and controlling DNA-reactive impurities, including nitrosamines. Manufacturers should have conducted thorough risk evaluations examining nitrosamine formation potential at every step of the API synthesis, formulation, and storage. For prazosin, this assessment should have flagged the secondary amine functional group as a particular concern and triggered enhanced control measures.
Analytical Method Development and Stability Testing: Sensitive analytical techniques such as liquid chromatography-tandem mass spectrometry (LC-MS/MS) and gas chromatography-mass spectrometry (GC-MS) can detect nitrosamines at nanogram-per-milliliter concentrations. These methods should have been developed not merely as compliance tools but as integral components of quality assurance. Accelerated and real-time stability studies should have characterized N-nitrosamine formation kinetics under various storage conditions, allowing manufacturers to project long-term accumulation and establish appropriate specifications.
Excipient Substitution and Formulation Optimization: Studies published over the past five years have clearly demonstrated that excipient selection significantly impacts nitrosamine formation risk. Replacing crospovidone with microcrystalline cellulose, substituting nitrocellulose blister packs with materials containing lower nitrite levels, and avoiding packaging materials that could leach nitrites would have substantially reduced formation potential. These are not expensive modifications—they are standard pharmaceutical quality optimization activities.
Use of Nitrite Scavengers: Ascorbic acid and alpha-tocopherol are established inhibitors of nitrosamine formation. These compounds react with nitrosating agents, preventing them from engaging with secondary amines. Incorporating these into formulations—a practice already established in regulatory guidance—provides an additional control layer. The cost of including these excipients is negligible compared to the business and public health costs of a recall.
Supply Chain Auditing: Incoming material testing should have included systematic monitoring of nitrite levels in all raw materials and excipients. Vendor qualification processes should have required suppliers to demonstrate nitrite control. Previous cases revealed that contamination could originate from recycled materials supplied by non-qualified vendors—a risk that rigorous supply chain management would have identified and eliminated.
For a medical device and pharmaceutical industry journalist, the most troubling aspect of the prazosin recall is that predictive signals existed in publicly available data streams, yet apparently went unheeded. Multiple early warning systems could have—and should have—triggered increased scrutiny of prazosin manufacturing before contaminated lots reached patients.
PubMed Literature Mining: Between 2020 and 2025, more than 30 peer-reviewed publications appeared in PubMed describing nitrosamine formation mechanisms, identifying affected drug classes, and proposing mitigation strategies. Publications from 2023-2024 specifically examined the role of excipient composition, crystal morphology, and storage conditions in driving nitrosamine formation. A systematic review of this literature would have identified prazosin—with its secondary amine functional group—as a compound warranting particular attention. Furthermore, publications describing the formation of N-nitrosamine impurities in APIs with similar chemical structures (such as other alpha-adrenergic agonists) should have triggered proactive risk assessments.
Trend Analysis in Pharmaceutical Recall Patterns: Research published in 2024 demonstrated that machine learning algorithms trained on PubMed citations and Google Trends data could predict pharmaceutical recalls with an AUC of 0.791 approximately 6 months to 12 months in advance. This research specifically examined how sudden spikes in search queries related to medication safety, combined with increased scientific literature citations, correlated with imminent recalls. For manufacturers and regulators monitoring such trend data, prazosin—given its secondary amine structure and the industry-wide focus on nitrosamines—should have appeared as a device with elevated risk scores.
FDA MedWatch and MAUDE Database Signal Detection: While the MAUDE database (Manufacturer and User Facility Device Experience) is typically associated with medical devices, the FDA's pharmacovigilance systems similarly aggregate adverse event reports for drugs. Careful analysis of adverse event patterns—such as clustering of complaints from specific geographic regions or manufacturing facilities, or temporal associations with specific manufacturing batches—can reveal early signals of quality problems. A 2017 study demonstrated that Internet search engine queries could predict drug recalls with approximately 6-fold enrichment when examined within specific geographic areas. Practitioners monitoring real-time pharmacovigilance signals should have potentially identified clusters of adverse events associated with prazosin batches before widespread distribution.
Regulatory Trend Analysis: The FDA's Early Alert program, expanded in September 2025 to encompass all medical devices, demonstrates the agency's commitment to shortening the gap between company awareness of safety issues and public notification. However, the program exists as a reactive tool. A more proactive approach would involve regulatory agencies systematically analyzing which drug categories, therapeutic classes, and chemical structures pose the highest nitrosamine formation risk based on current manufacturing practices and recent recalls. Prazosin, given its structural similarity to compounds already recalled for nitrosamine contamination, should have been flagged for enhanced surveillance and manufacturer audits.
The prazosin recall reveals several interconnected systemic failures:
Inadequate Risk Stratification: Not all drugs are equally vulnerable to nitrosamine formation. Yet regulatory guidance and manufacturer responses have often treated the problem as if all secondary amine-containing drugs require identical control measures. A more differentiated approach would have recognized that prazosin—a chronic-use medication with a vulnerable secondary amine structure—warranted heightened scrutiny and potentially enhanced specifications compared to shorter-course therapies or compounds with less reactive amine groups.
Delayed Implementation of Known Control Strategies: The prazosin recall occurred despite seven years of industry knowledge regarding nitrosamine risks and published mitigation strategies. This suggests that regulatory compliance and risk management responses have lagged behind scientific understanding. Manufacturers should not require recalls to motivate implementation of preventive measures already documented in peer-reviewed literature and regulatory guidance.
Fragmented Surveillance Systems: Different regulatory agencies, manufacturers, and research institutions maintain separate databases and monitoring systems. While the FDA's recall database, PubMed, Google Trends, and industry trend reports exist in parallel, no unified system automatically flags emerging patterns. A manufacturer monitoring only its own internal complaint data and FDA enforcement actions might miss trend signals visible across multiple data streams.
Insufficient Supply Chain Integration: The contamination may originate not with Teva's manufacturing process but with upstream suppliers of the API, excipients, or manufacturing reagents. Yet many pharmaceutical companies maintain arms-length relationships with suppliers, conducting limited audit and testing. Enhanced supply chain integration—including vendor audits specifically targeting nitrosamine formation risk factors—represents a preventive investment that many manufacturers have not fully embraced.
For a medical device industry analyst, the prazosin recall suggests that a more sophisticated predictive framework could have prevented this contamination from reaching patients. Such a framework would integrate multiple data streams:
Structural Chemical Profiling: Systematically categorize all marketed drugs by their chemical structure's inherent vulnerability to nitrosamine formation. Use quantitative structure-activity relationship (QSAR) models—well-established tools in pharmaceutical chemistry—to identify high-risk compounds. Flag prazosin and similar secondary amine-containing drugs for enhanced monitoring.
Manufacturing Process Audits: Conduct periodic comprehensive audits of manufacturing facilities not merely for compliance with current regulations but for adherence to best practices described in recent scientific literature. Specifically, verify that manufacturers are implementing LC-MS/MS testing, conducting proper stability studies, and using excipients known to have lower nitrite content.
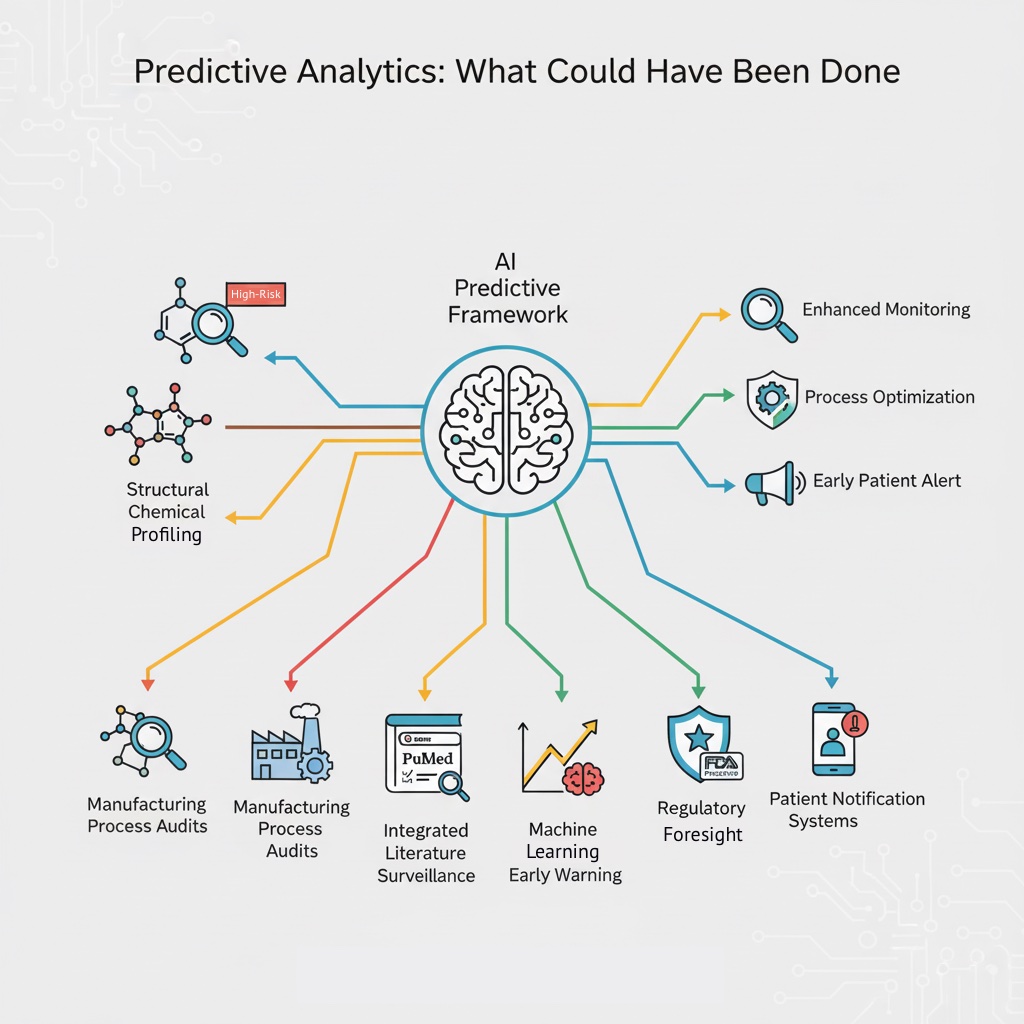
Integrated Literature Surveillance: Establish automated systems that continuously scan PubMed, regulatory databases, and industry publications for emerging safety signals related to drug classes or manufacturing processes. Use natural language processing to identify when new research identifies vulnerability factors applicable to marketed drugs. Alert manufacturers when literature identifies risk factors potentially present in their products.
Machine Learning-Based Early Warning: Implement the machine learning approaches demonstrated in 2024 research, training algorithms on combinations of PubMed data, Google Trends, FDA recall history, and manufacturer-specific variables to generate probabilistic recall risk scores. Use these scores to trigger enhanced surveillance, audits, or voluntary reformulation efforts before recalls become necessary.
Regulatory Foresight: Move from reactive recalls to proactive regulatory engagement. The FDA could establish a "high-risk drug" category for secondary and tertiary amine-containing drugs, requiring manufacturers to implement enhanced specifications, more frequent testing, and premarket stability data before approval. Such an approach would shift the burden of prevention onto manufacturers rather than relying on post-market detection.
Patient Notification Systems: Implement more responsive systems for notifying patients and healthcare providers when a drug is identified as high-risk for contamination, even before a formal recall. Given the prazosin population—many taking the drug chronically for PTSD—early notification would allow for alternative medication transitions and reduce disruption.
The prazosin hydrochloride recall is not a surprise. It represents a predictable consequence of manufacturing process gaps that should have been identified and corrected years ago. The scientific literature has provided clear guidance on nitrosamine formation mechanisms since 2018. Analytical methods to detect these impurities exist and are well-documented. Control strategies—excipient substitution, pH management, nitrite scavengers, supply chain auditing—are not novel; they are standard pharmaceutical quality optimization practices.
What failed in this case is not science or technology but organizational will and integrated risk management. Manufacturers did not conduct sufficiently rigorous risk assessments. Regulatory agencies did not establish differentiated oversight based on chemical structure vulnerability. And industry surveillance systems remained fragmented rather than integrated to capture emerging trends.
For patients taking prazosin, the recall creates uncertainty and potential disruption to their mental health treatment. For healthcare providers, it erodes confidence in pharmaceutical quality. For manufacturers, it represents a costly recall affecting over 500,000 units—a consequence that proper preventive management would have easily justified.
The pharmaceutical industry and its regulators must move beyond reactive recalls toward predictive prevention. The tools exist: scientific literature surveillance, machine learning-based risk modeling, structural chemical analysis, and integrated pharmacovigilance databases. What remains is the commitment to deploy these tools systematically, transparently, and before patients are harmed.

Explore all features of PMM for 2 weeks to see how it can simplify your post-market surveillance. If you cancel before the trial ends, your credit card will not be charged

.png)





