By
Irina Smith
August 4, 2025
•
15
min read

Two deaths and 79 serious injuries linked to a recently cleared medical device paint a sobering picture of what happens when early warning signs go unheeded. The recall of Baxter's Novum IQ large-volume infusion pump in July 2025—just 15 months after receiving FDA clearance—raises critical questions about how the medtech industry identifies, predicts, and prevents device failures before they claim lives.
The incident involves more than regulatory compliance or corporate liability. It represents a systemic failure to integrate multiple data streams that, when analyzed collectively, revealed clear predictive signals of impending catastrophe. From FDA adverse event databases to peer-reviewed scientific literature and frontline clinical feedback on social media, the evidence was there. The question is: why wasn't it enough?
Baxter International received FDA clearance for its Novum IQ large-volume infusion pump with Dose IQ safety software in April 2024. The device was marketed as a technological advancement, featuring a web-based customizable drug library, dose error reduction system, and titration error prevention technology designed to provide additional safety measures. The company positioned the platform as "redefining the future of infusion therapy" and touted its integration capabilities with electronic medical records.

The first cracks in this promising narrative appeared quickly. By June 2024, just two months after launch, Baxter issued an urgent medical device correction addressing potential underinfusion following use of the "standby mode" feature. Testing revealed that at flow rates above 50 mL/hour, certain infusions experienced flow rate variability exceeding 10% after the pump remained in standby mode for 2 hours and 30 minutes. In worst-case scenarios, 50% underinfusion could occur at the maximum flow rate of 1,200 mL/hour after the maximum standby time of 12 hours. This initial recall was associated with one serious injury and no deaths.
The situation deteriorated dramatically by July 2025. Baxter issued a second urgent medical device correction, this time involving two distinct but related issues. First, the pump demonstrated a propensity for underinfusion when transitioning from a lower flow rate to a higher one—specifically when the second rate exceeded double the first rate. The level of underinfusion varied based on how long the pump operated at the lower rate and the magnitude of the rate change. In the worst case, no drug delivery occurred at all.
Second, Baxter identified an increase in customer reports of over- and underinfusion potentially caused by set misloading. When tubing was not properly loaded into the pump channel, the device infused at rates higher or lower than programmed. The consequences ranged from minor harm to death, depending on the patient's condition, the medication being administered, and the clinical context. High-risk patients faced serious adverse health consequences including hemodynamic instability, cardiac arrhythmias, insufficient sedation, hyperglycemia, and thromboembolic events.
As of June 27, 2025, Baxter reported 79 serious injuries and two deaths potentially associated with these issues. The FDA issued an early alert under its recall communications pilot program, classifying this as a Class I recall—the most serious type.
The Novum IQ recall reveals multiple layers of failure spanning device design, human factors engineering, and software validation. Understanding these root causes is essential to comprehending how such a catastrophic failure occurred so soon after regulatory approval.
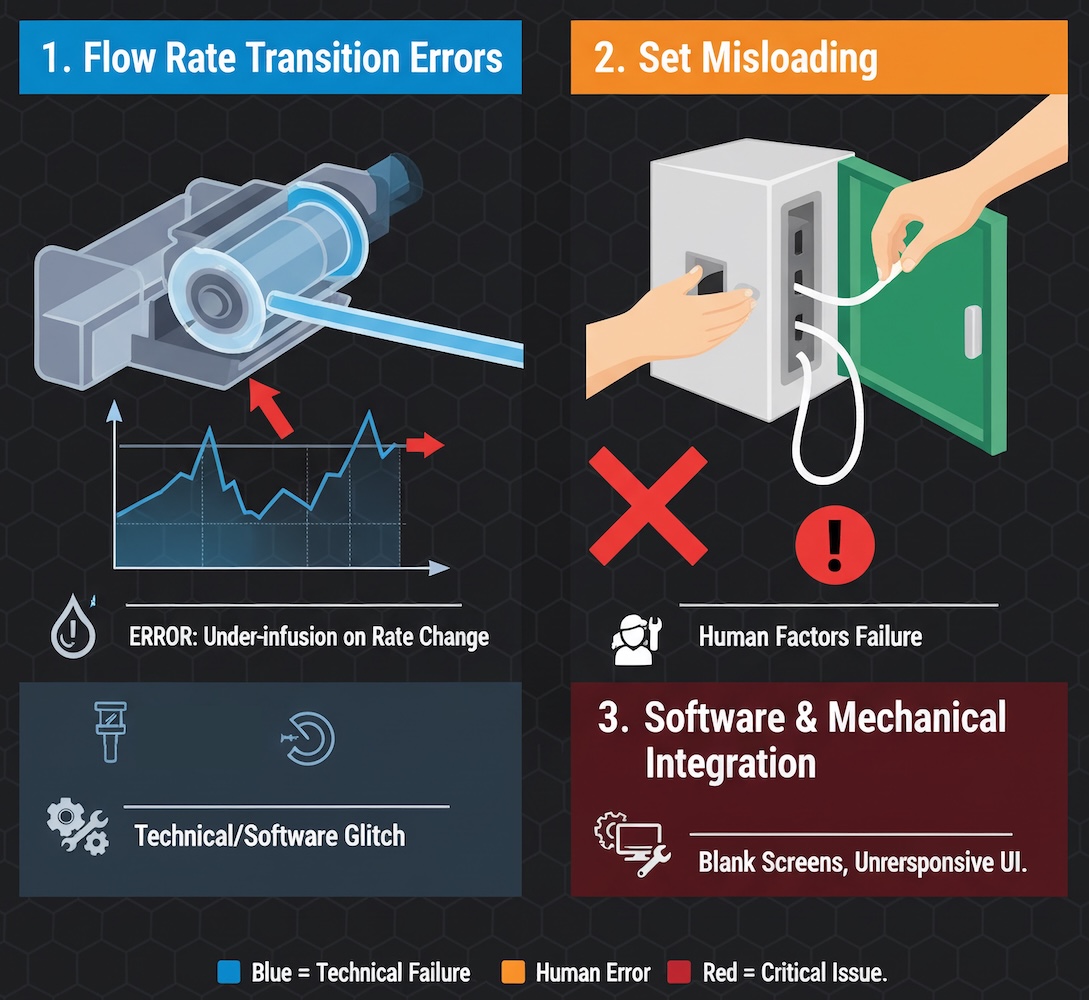
Flow Rate Transition Errors
The most technically complex issue involves the pump's inability to accurately deliver medication when transitioning between flow rates. When a clinician programs a significant rate increase—for example, moving from a 30 mL/hour primary infusion to a 600 mL/hour bolus—the pump fails to deliver the intended volume. This problem intensifies with longer durations at the lower rate and larger magnitude rate changes. The mechanical explanation likely involves tubing compression and the pump's peristaltic mechanism failing to properly "catch up" when demands suddenly increase.
This issue is not novel to infusion pump technology. A 2025 study applying high-reliability principles to infusion pump safety identified 112,875 upstream occlusion events for 389,604 infusion starts—a failure rate of 29% within six months. The research emphasized that flow rate accuracy is affected by multiple variables including tubing characteristics, medication viscosity, and infusion rates. Yet Baxter's design apparently failed to account for these well-documented phenomena during development and validation.
Set Misloading: A Predictable Human Factors Failure
The second major issue—tubing misloading—represents a classic human factors engineering failure. When the pump door is not fully opened before loading, or when tubing is not loaded taut and without slack in the pumping channel, the device infuses at incorrect rates. This design flaw allows users to complete what appears to be proper setup while the device operates in a fundamentally unsafe state.
Human factors research in infusion pump safety has documented this type of failure extensively. A 2006 study specifically identified "tubing misload allows free flow event with smart intravenous infusion pump". A 2019 hazard analysis of Class I infusion pump recalls found that inaccurate dosage delivery—either over- or underdelivery of fluid—was a critical source of product technical risk. The analysis identified energy hazards and product component failures as major hazard forms associated with infusion pumps.
A 2025 review of human factors and infusion pumps noted that a high percentage of sentinel/drug-adverse events result from pump use errors. The review emphasized that designing infusion pumps requires careful attention to user interface design, workflow integration, and user training. Studies have shown that when infusion pumps are properly designed with human factors principles, they can prevent errors in almost 1 case per 1,000 admissions.
The Novum IQ's misloading vulnerability suggests inadequate human factors validation during the design phase. FDA guidance requires medical device manufacturers to conduct formative and summative human factors studies to identify and mitigate use errors. The fact that this fundamental loading error escaped detection indicates either insufficient testing with representative users in realistic clinical scenarios, or a failure to act on findings from such testing.
Software and Mechanical Integration Failures
Beyond the standby mode issue, Baxter issued additional alerts in August 2025 regarding software anomalies causing blank run screens. While these screens do not interrupt ongoing therapy, they prevent users from adjusting treatments—a potentially critical limitation in dynamic clinical situations. The proliferation of software-related issues so soon after launch suggests rushed development timelines and insufficient validation testing.
Perhaps the most troubling aspect of the Novum IQ recall is that it was eminently predictable. Multiple data sources provided clear warning signals that, if properly analyzed and acted upon, could have prevented injuries and deaths.
FDA MAUDE Database: The Canary in the Coal Mine
The FDA's Manufacturer and User Facility Device Experience (MAUDE) database contains adverse event reports that, when analyzed systematically, can reveal patterns predictive of major device failures. In the case of the Novum IQ, MAUDE reports from February through May 2025 documented the exact problems that would culminate in the July recall.
A February 2025 report described a Novum IQ over-infusing fentanyl at a rate "much higher than programmed," with the patient receiving up to three times the ordered rate. An April 2025 report documented severe underinfusion: a pump programmed to deliver 500 mL at 999 mL/hour actually delivered only 100 mL/hour. When reprogrammed, the same issue recurred. A May 2025 report described over-infusion where a 100cc bag of midazolam programmed at 25 cc/hour completed in 2 hours instead of the expected 4 hours.
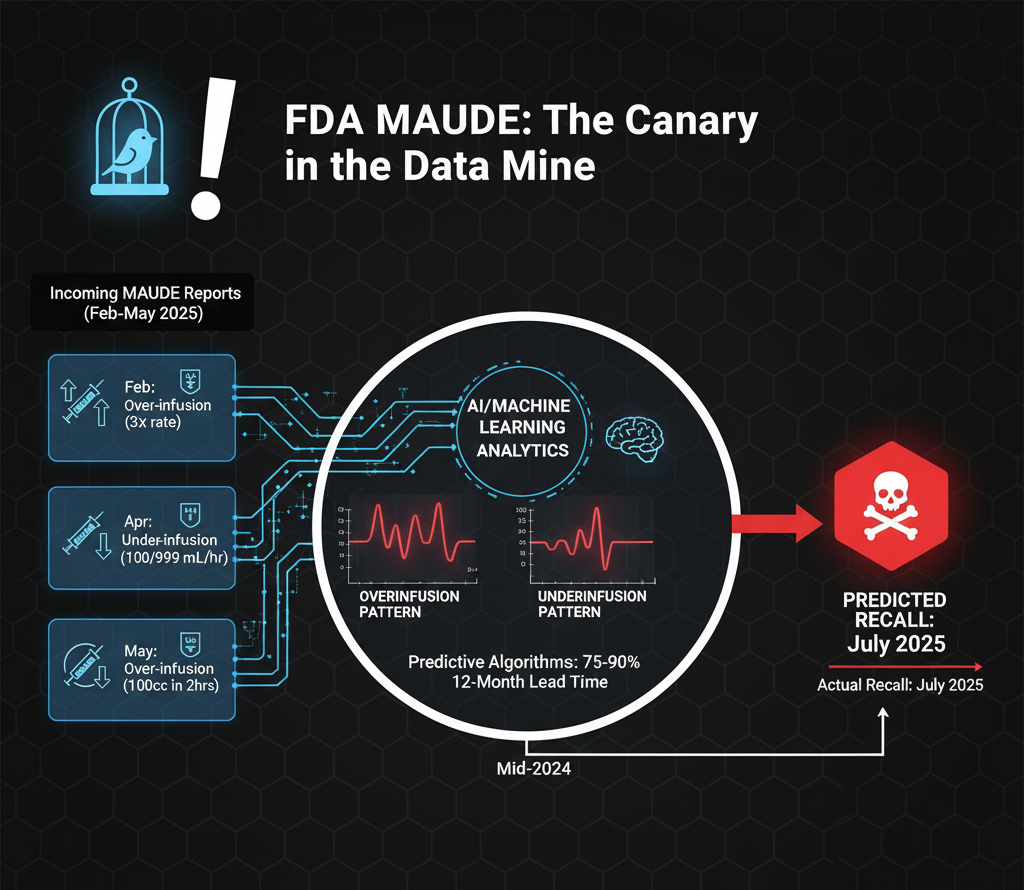
These reports demonstrate both over- and underinfusion patterns—the exact dual failure modes later cited in the recall. While individual reports showed "no clinical signs, symptoms or conditions" or "no injury reported," the pattern across multiple reports should have triggered immediate investigation. The discrepancy between individual reports claiming no injury and the FDA's eventual tally of 79 serious injuries and 2 deaths suggests systematic underreporting in the MAUDE system itself.
A 2024 machine learning study demonstrated that algorithms using publicly available data could predict FDA medical device recalls with sensitivities of 75-90% and specificities of 100%, with lead times as great as 12 months. The study specifically noted that infusion pumps were among the devices most amenable to predictive analytics due to their ubiquity and the volume of adverse event data generated. Had Baxter or the FDA employed such analytics, the July 2025 recall could have been predicted as early as mid-2024.
Scientific Literature: A Roadmap of Known Failure Modes
The scientific literature on infusion pump safety reads like a preview of the Novum IQ's problems. A 2010 FDA Infusion Device Summit highlighted 56,000 reported incidents related to intravenous infusions, resulting in 710 deaths. The conference emphasized that a high percentage of sentinel events result from pump use errors and stressed the need for improved safety features and user protocols.
More recent studies have confirmed these issues persist. Research published in 2018 found that using infusion pumps leads to errors in up to 60% of infusions, with problems including incomplete labeling, incorrectly marked systems, infusion rate errors, and incorrect dosages. A 2021 study analyzing smart pump event logs found that 28.7% of all unique infusions had at least one operational alarm, with 8% of alarms taking over 4 minutes to resolve.
Studies on flow rate accuracy have documented how multiple variables affect pump performance. A 2023 systematic review found that flow rate accuracy is influenced by infusion device position, fluid viscosity, tubing characteristics, and environmental factors. Research has shown that at low flow rates (below 1 mL/hour), accuracy can vary significantly, and that transitions between flow rates create particular vulnerability to dosing errors.
The literature on set misloading is equally prescient. Studies on multi-infusion therapy have documented "spaghetti syndrome" where multiple IV lines become entwined, leading to mix-up errors and delays. Research on infusion identification found that when tubing is improperly organized or loaded, clinicians struggle to reconcile bags with associated tubing, pumps, and patient access ports, resulting in errors that have caused patient harm and death.
This body of knowledge should have informed every stage of the Novum IQ's development. The failure to design around these well-documented vulnerabilities represents either ignorance of the scientific literature or a conscious decision to prioritize time-to-market over patient safety.
Social Media and Clinical Forums: The Voice of Frontline Users
While MAUDE reports and scientific literature provide formal documentation, social media and clinical forums offer real-time, unfiltered feedback from frontline clinicians. These informal channels have long served as early warning systems for device problems, yet they remain largely untapped by manufacturers and regulators.
A Reddit discussion from November 2021 titled "Why are so many nurses hating on Baxter pumps?" provides revealing insight into longstanding issues with Baxter infusion devices. Nurses described Baxter pumps as "inaccurate pieces of trash" and noted that "there is a huge recall of hundreds of thousands of devices due to flow rate accuracy back in 2010". One critical care nurse explained: "They are inaccurate as hell depending on different variables like IV gauges and viscosity of medication. I called them out on it... conducted very controlled experiments... the volume delivered [doesn't match] the volume entered".
The nurse continued: "Despite video evidence to the contrary... Baxter, of course, deny[s] it all. Then we're all crazy apparently". This account suggests that clinicians were reporting accuracy problems to Baxter well before the FDA clearance of the Novum IQ, yet the company dismissed these concerns.
A 2022 post on a medical device safety forum discussed the Baxter Sigma Spectrum infusion pump's rate accuracy of +/-5%, noting that "over the course of extended infusions (12-24 hours), a +5% variability can cause the infusion to end up to 2 hours early". This observation directly foreshadows the Novum IQ's flow rate variability issues.
These discussions reveal a pattern: Baxter has struggled with flow rate accuracy across multiple product lines for over a decade. The 2010 recall for the Colleague pump involved the same fundamental issue—inaccurate flow rates—yet the company apparently failed to learn from this experience when developing the Novum IQ.
Historical Context: The Baxter Colleague Recall of 2010
The Novum IQ recall is not an isolated incident but rather the latest chapter in Baxter's troubled history with infusion pump accuracy. In 2010, the FDA identified infusion pump safety as a critical priority, citing 56,000 adverse event reports and 87 pump recalls in the preceding five years. Baxter's Colleague pump was among those recalled for flow rate accuracy issues.
A biomedical engineering forum from 2002 documented concerns about Baxter Colleague pumps failing rate accuracy checks during routine service, with technicians noting that "the longer the pump has been running the greater the error percentage the pump under infuses". This suggests a fundamental mechanical wear issue that Baxter never fully resolved.
The fact that Baxter encountered similar flow rate problems with the Colleague pump in 2010, again with the Sigma Spectrum pump in subsequent years, and now with the Novum IQ in 2024-2025 indicates a systematic corporate failure to address root causes. Each generation of pumps appears to repeat the mistakes of its predecessors, suggesting inadequate institutional learning and quality systems.
Preventing recalls like the Novum IQ requires a comprehensive, data-driven approach that integrates multiple surveillance streams and acts decisively on early warning signals.

Enhanced Pre-Market Validation
The FDA's 510(k) clearance pathway, under which the Novum IQ was approved, requires manufacturers to demonstrate substantial equivalence to a predicate device. However, substantial equivalence does not guarantee safety or effectiveness—it merely establishes that a new device is not substantially different from an already-marketed device. This pathway has been criticized for allowing devices with incremental design changes to bypass the more rigorous pre-market approval (PMA) process.
For high-risk devices like infusion pumps, more extensive pre-market testing should include:
Real-Time MAUDE Analytics
The FDA should implement machine learning algorithms to continuously monitor MAUDE reports for early signals of device problems. Studies have demonstrated that such systems can predict recalls with high accuracy and substantial lead times. Key features of an effective system would include:
A 2024 study on artificial intelligence-related safety issues in medical devices demonstrated that AI/ML algorithms can effectively analyze patient safety event reports to identify how device features contribute to safety issues. Applying these techniques systematically to MAUDE data could transform post-market surveillance from reactive to proactive.
Social Listening and Clinician Feedback Loops
Manufacturers and regulators should systematically monitor social media, clinical forums, and professional networks for early signals of device problems. While informal, these channels often provide the earliest warnings of safety issues. A structured social listening program would include:
The Novum IQ case demonstrates the cost of ignoring frontline user feedback. Nurses were reporting Baxter pump accuracy problems for years, yet these concerns apparently did not influence the Novum IQ's design or validation.
Independent Post-Market Surveillance Studies
Given the limitations of manufacturer-reported MAUDE data, independent researchers and healthcare systems should conduct prospective surveillance studies of high-risk devices. A 2025 study applying high-reliability principles to infusion pump safety demonstrates the value of such efforts. The researchers identified 112,875 upstream occlusion events across a multi-site health system in just six months—a failure rate that would never have been apparent from MAUDE reports alone.
Healthcare systems implementing new infusion pumps should:
Regulatory Reform
The Novum IQ recall exposes fundamental weaknesses in the FDA's regulatory framework for medical devices. Reform should include:
The Baxter Novum IQ recall offers several critical lessons for medical device manufacturers:

First, institutional memory matters. Baxter's repeated struggles with infusion pump flow rate accuracy across multiple product generations—Colleague (2010), Sigma Spectrum (ongoing), and now Novum IQ (2024-2025)—suggest a failure to learn from past mistakes. Organizations must establish robust quality management systems that capture lessons from previous failures and systematically apply them to new product development.
Second, listen to frontline users. The nurses who reported accuracy problems with Baxter pumps for years were not "crazy" as one user was told. They were providing valuable real-world feedback that, if heeded, could have prevented the Novum IQ's problems. Manufacturers must establish formal mechanisms to capture, evaluate, and act on clinician feedback—even when it contradicts internal test data.
Third, prioritize safety over time-to-market. The rapid progression from FDA clearance (April 2024) to first recall (June 2024) to catastrophic recall with deaths (July 2025) suggests insufficient validation testing before market release. While competitive pressures drive aggressive product launch timelines, the reputational and financial costs of a major recall far exceed any short-term market advantage.
Fourth, embrace transparency. When device problems emerge, manufacturers should engage openly with clinicians, regulators, and patients rather than denying issues. Baxter's apparent dismissal of nurse concerns about pump accuracy ultimately undermined trust and may have delayed identification of the Novum IQ's problems.
Two deaths and 79 serious injuries represent not just statistics but preventable human tragedies. Each patient harmed by the Novum IQ's failures trusted that the medical device industry and regulatory system would protect them. That trust was betrayed by a confluence of failures: inadequate design, insufficient validation, poor post-market surveillance, and institutional blindness to warning signals that were clearly visible across multiple data sources.
The path forward requires fundamental transformation in how medical devices are developed, evaluated, and monitored. Predictive analytics must evolve from research curiosity to operational imperative. MAUDE data, scientific literature, and social media feedback must be integrated into comprehensive surveillance systems that identify problems before they cause harm. Manufacturers must prioritize patient safety over competitive advantage, and regulators must enforce accountability with meaningful consequences for failures.
Most importantly, the voices of frontline clinicians—the nurses who operate infusion pumps thousands of times per shift, who intuitively recognize when devices behave abnormally—must be elevated from background noise to critical signal. Their observations, shared informally on Reddit and nursing forums, provided years of advance warning about Baxter pump problems that formal surveillance systems failed to detect.
The Novum IQ recall proves that we possess the data and analytical tools to predict device failures before they claim lives. What we currently lack is the institutional will to act on those predictions decisively and courageously. Until that changes, tragedies like this will continue to unfold with depressing regularity, each one entirely preventable in retrospect, each one needlessly devastating to those who trusted the system to protect them.

Explore all features of PMM for 2 weeks to see how it can simplify your post-market surveillance. If you cancel before the trial ends, your credit card will not be charged

.png)