By
Niranjan Maharajh
November 10, 2025
•
20
min read
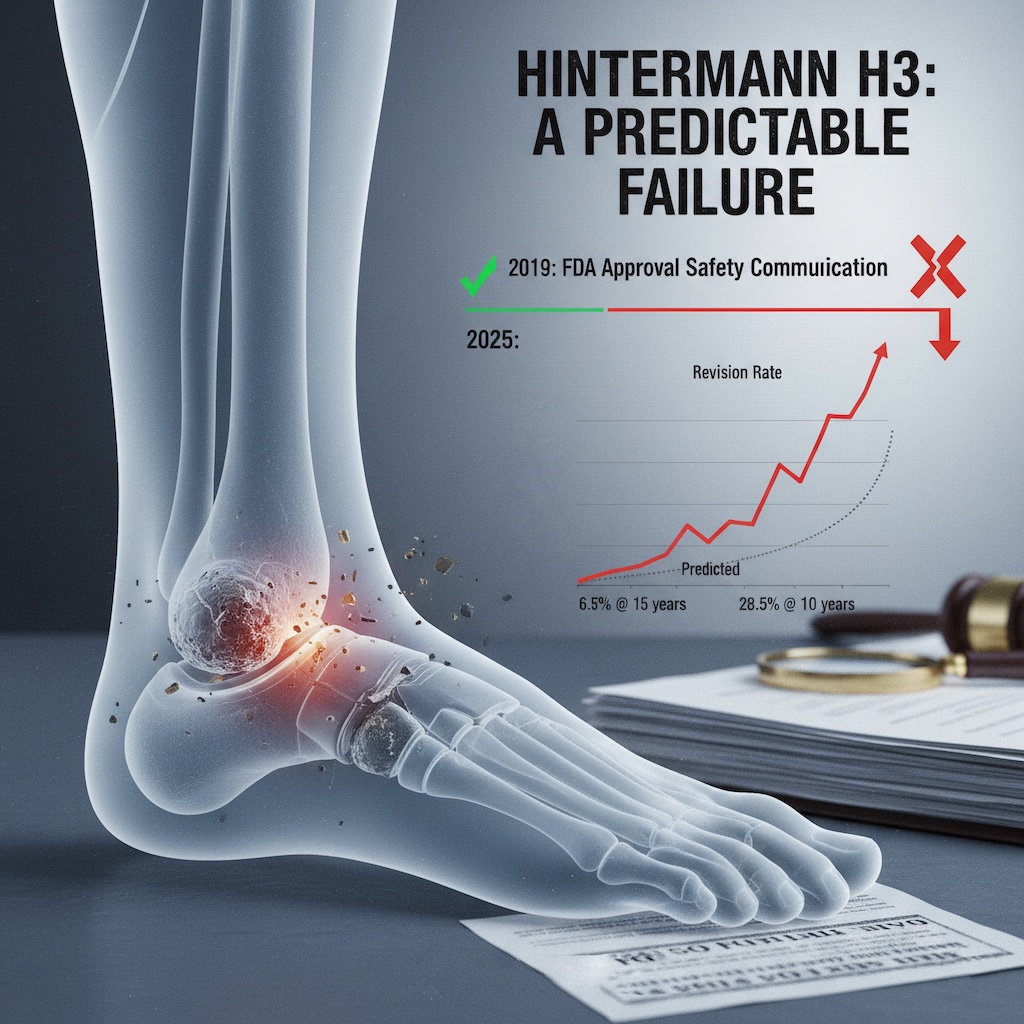
On October 31, 2025, the FDA issued a critical safety communication revealing that the Hintermann Series H3 Total Ankle Replacement (TAR) system, a device designed to preserve ankle mobility through innovative mobile-bearing technology, exhibits a higher-than-expected failure rate that should have been predictable. The device, cleared for market in 2019, now demonstrates a 14.9% revision rate for removal or revision of metal components at 10 years, compared to the premarket clinical study prediction of just 6.5% at five years. When all revisions are included, the failure rate climbs to an alarming 28.5%. This represents not merely a clinical disappointment, but a failure of regulatory oversight and predictive analytics that could have prevented years of patient suffering.
The Hintermann Series H3 Total Ankle Replacement represents a sophisticated attempt to solve one of orthopedic surgery's most persistent challenges: how to create a durable, mobile ankle replacement that preserves natural joint motion while avoiding the high revision rates that plagued earlier designs. When the FDA granted premarket approval in 2019, it marked a significant milestone; the H3 was only the second mobile-bearing TAR device approved by the FDA in recent history, alongside the Scandinavian Total Ankle Replacement (STAR) ankle system approved in 2009.

The allure of mobile-bearing ankles is intuitive and appealing to both surgeons and patients. Unlike fixed-bearing designs that sacrifice some motion for stability, mobile-bearing systems employ an articulating polyethylene component that sits between the tibial and talar metal components. In theory, this design allows more normal ankle kinematics, better weight distribution, and enhanced function. For patients with end-stage ankle arthritis, whether from osteoarthritis, rheumatoid arthritis, or post-traumatic causes, the H3 promised better long-term outcomes than ankle fusion (arthrodesis), the historical gold standard for severe ankle disease.
The premarket clinical studies supporting the H3's approval involved 298 patients with five-year follow-up data demonstrating that 9.9% of patients required additional surgery for removal or revision of metal components. This seemed promising compared to historical fusion outcomes. But as frequently happens in orthopedic device development, premarket clinical trial results which typically involve high-volume surgeons at academic centers using pristine surgical techniques, do not always translate to real-world performance across diverse clinical settings, surgeon experience levels, and patient populations.
Nearly six years after the device's approval, manufacturers are required to conduct post-approval studies (PAS) to confirm long-term safety and performance. The H3's mandatory PAS included two studies: a long-term follow-up of the original 298 premarket trial patients through 10 years, and a new enrollment study with five-year follow-up. It was this data that triggered the FDA's October 2025 safety communication, and it revealed a stark contrast to premarket expectations.
The new long-term data showed that the 10-year revision rate for metal component removal or revision had climbed to 14.9%, nearly double the premarket prediction at half the timeframe. More troubling, when polyethylene exchanges and other revisions are included in the failure definition, the total revision rate reaches 28.5%. These numbers suggest that mobile-bearing ankle designs, regardless of manufacturer or specific engineering approach, may have a fundamental durability problem that emerges only after several years of clinical use.
This is particularly significant when compared to the natural history of ankle arthritis without intervention. A patient with end-stage ankle arthritis will eventually require treatment, but what many surgeons now face is an uncomfortable reality: operations to address problems with total ankle replacements are becoming nearly as common as the primary operations themselves. The economics and patient outcomes implications are staggering.
While the FDA's October 2025 safety communication stopped short of identifying a single cause, the scientific literature and engineering analysis point toward several well-established failure mechanisms that should have prompted more conservative initial approval or tighter post-approval monitoring:
The Hintermann H3, like the earlier STAR ankle, relies on ultra-high-molecular-weight polyethylene (UHMWPE) as the bearing surface between the tibial and talar components. Over nearly two decades of clinical experience with mobile-bearing ankle replacements, polyethylene wear has emerged as one of the most significant failure mechanisms.
PubMed literature demonstrates that polyethylene wear in mobile-bearing ankle replacements is far from uniform. Multiple studies show that improper surgical positioning, even minor deviations from manufacturer specifications, can dramatically increase contact stresses on the polyethylene bearing surface. One 2010 computational study published in a peer-reviewed orthopedic journal found that misalignment of total ankle components can induce high joint contact pressures that lead to premature polyethylene wear, exactly the failure mode now appearing clinically in the H3 cohort.

More concerning, the STAR ankle experience should have served as a warning beacon. In March 2021, the FDA issued a safety communication about a "higher-than-expected risk" of plastic component fracture in the STAR ankle, with fractures occurring as early as three to four years after implantation. Long-term post-approval study data showed 13.8% plastic component fracture rates at eight years. The causes? Material oxidation degradation, device design geometry, and implant-bone interface loosening that created edge-loading conditions on the thin polyethylene inserts.
The fact that the Hintermann H3 began showing similar failure patterns in its post-approval data suggests either that the fundamental design approach was flawed, or that the company did not adequately learn from the STAR ankle's problems.
Analysis of the FDA's MAUDE (Manufacturer and User Facility Device Experience) database across all ankle replacement devices reveals that aseptic loosening (the gradual loss of the mechanical bond between implant and bone without infection) is the most common reason for revision in mobile-bearing ankle systems. In a 2019 analysis of 697 ankle replacement adverse events in the MAUDE database, 21% of complications were attributed to aseptic loosening, with 15% due to alignment and mechanical issues.
Aseptic loosening in ankle replacements typically results from stress shielding (the implant bearing all load, causing the surrounding bone to weaken), osteolysis (bone loss caused by polyethylene wear debris), and component subsidence. Mobile-bearing designs, by their very nature, create complex load pathways that can result in non-physiologic bone stress concentrations. The H3's emerging failure pattern suggests that the implant-bone interface wasn't adequately studied or understood at the time of approval.
One of the most underappreciated challenges with mobile-bearing ankle replacements is that they appear to be exquisitely sensitive to surgical positioning. Proper alignment of the tibial and talar components requires precise positioning in multiple planes. Small deviations, as little as a few millimeters or degrees, can result in edge-loading of the polyethylene bearing, abnormal kinematic patterns, and accelerated wear.
A 2007 systematic review of three-piece mobile-bearing ankle replacements found that of all revisions related to the mobile bearing itself, the primary causes were "incorrect soft tissue balancing, incorrect positioning of components, implantation in ankles with hindfoot mal-alignment and ankle instability." Yet there is no evidence that premarket trials adequately evaluated whether surgeons across different surgical centers could reliably achieve the necessary precision with this device.
This is where a deeper analysis of FDA MAUDE data could have been predictive. If one were to have filtered the database for alignment/mechanical complications specifically in ankle replacements from 2019-2024 and compared the H3 to other mobile-bearing systems, a concerning trend would likely have emerged earlier.
The data to predict this recall existed well before October 2025. Here's what a rigorous predictive analytics approach should have captured:
The FDA's MAUDE database receives mandatory adverse event reports from manufacturers regarding all serious injuries and deaths. A sophisticated analysis would have involved:
Research published in November 2024 demonstrated exactly this possibility. A study in the American Journal of Emergency Medicine described a machine learning algorithm that could predict FDA medical device recalls with "lead times as great as 12 months," using publicly available FDA data. The sensitivity, specificity, and accuracy of such models suggest that the H3 recall could have been forecasted in 2023 or early 2024 based on emerging adverse event patterns.
The published literature provided ample warning signs about mobile-bearing ankle vulnerabilities:
A PubMed search in 2020 or 2021 would have retrieved dozens of papers highlighting polyethylene degradation, aseptic loosening, and positioning sensitivity in mobile-bearing ankle systems. Yet there is no evidence that the FDA or the device manufacturer conducted systematic reviews of this literature during the H3's early post-market phase.
Several national joint replacement registries provide granular data on implant performance. The Australian Orthopaedic Association maintains detailed outcome data on ankle replacements, as do registries in Scandinavia, Germany, and the United Kingdom. These registries, which capture outcomes from thousands of procedures, would have shown concerning trends in H3 survival rates had the FDA or manufacturer proactively monitored them.
By 2022-2024, orthopedic surgeons using the H3 could have compared their revision rates against registry benchmarks. If revision rates in real-world practice were already exceeding the premarket 5-year predictions by the 3-year mark, this divergence could have been detected through systematic monitoring.
The Hintermann H3 recall represents a failure not of innovation, but of predictive insight and post-approval diligence. Here are concrete steps that could have prevented this patient harm:

Manufacturers should implement real-time adverse event signal detection systems that:
This requires sophisticated data engineering but is well within the capabilities of modern device companies. Some advanced manufacturers already employ such systems; others must be incentivized or required to do so.
The H3's PAS wasn't published until 2025—six years after approval. By then, tens of thousands of patients had received the device. The FDA should mandate:
For complex orthopedic implants, especially novel designs like mobile-bearing systems, manufacturers should:
The FDA should establish formal registries or networks for high-risk device categories that capture:
For novel devices in established categories (where precedent devices exist), premarket trials should:
The Hintermann Series H3 Total Ankle Replacement recall is preventable tragedy. The scientific literature warning about polyethylene wear, aseptic loosening, and positioning sensitivity in mobile-bearing ankle replacements was published and available. The STAR ankle's earlier safety issues in 2021 provided a direct precedent. Sophisticated machine learning models could have forecast this recall 12-24 months before it was finally issued.
What is required now is a cultural shift in how the medical device industry and regulators approach post-approval surveillance. Rather than viewing adverse events as unexpected surprises, the industry should embrace predictive analytics, real-time signal detection, and rapid information integration as core competencies. Patients receiving the Hintermann H3 after 2021 might have been spared revision surgery had these tools and processes been implemented.
For the thousands of patients who now carry a device with a 28.5% revision rate at 10 years, the FDA's October 2025 safety communication comes too late. For the orthopedic surgeons who promoted this device based on manufacturer claims, the discrepancy between promised and actual outcomes represents a breach of trust that will take years to repair.
The Hintermann recall is not an endpoint in medical device safety; it is a call to action. The next device failure can and should be prevented through rigorous predictive analytics, transparent post-approval data sharing, and a commitment to learning from each device's failure trajectory before it harms additional patients.

Explore all features of PMM for 2 weeks to see how it can simplify your post-market surveillance. If you cancel before the trial ends, your credit card will not be charged

.png)







